A prion is a misfolded protein that can trigger normal proteins in the brain to misfold and accumulate, leading to progressive neurodegenerative disorders known as prion diseases. Prions are unique pathogens as they are composed solely of protein and lack genetic material like DNA or RNA. The normal cellular prion protein (PrPC) can be converted into its pathogenic form (PrPSc) through a process that alters its folding pattern, typically changing an alpha helix into a beta sheet conformation. This misfolded form can then interact with and convert other normal prion proteins, leading to a chain reaction of protein misfolding and aggregation. Prion diseases, also called transmissible spongiform encephalopathies (TSEs), can occur sporadically, be inherited, or acquired through infection, affecting both humans and animals. The accumulation of misfolded prion proteins in the brain leads to neuronal death, resulting in characteristic spongiform changes and various neurological symptoms.
Characteristics of Prions
Prions are unique infectious agents with several distinctive characteristics:

1. Protein-based infectious agents
Prions are unique infectious agents composed solely of misfolded proteins, specifically the prion protein (PrP). Unlike conventional pathogens that contain nucleic acids, prions propagate by inducing normal cellular prion proteins (PrPC) to adopt the misfolded, pathogenic conformation (PrPSc). This protein-only nature of prions challenges traditional concepts of infectious agents and has significant implications for understanding the mechanisms of disease transmission and pathogenesis. The prion hypothesis, which posits that the infectious agent is an aberrant conformational isoform of PrP, has been substantiated by experiments demonstrating that recombinant PrP can induce prion disease in animal models`1.
2. Self-propagating
Prions propagate by a process known as seeded aggregation, where the misfolded prion protein (PrPSc) acts as a template to convert normal prion proteins (PrPC) into the misfolded form. This self-propagating mechanism allows prions to spread within the host and between individuals, leading to the progressive accumulation of PrPSc in the brain and other tissues. The ability of prions to self-replicate without nucleic acids is a defining feature that distinguishes them from other infectious agents and has broadened our understanding of protein misfolding diseases.
3. Neurodegenerative effects
Prion diseases are invariably fatal neurodegenerative disorders characterized by the progressive loss of neuronal cells and the accumulation of misfolded prion proteins in the brain. These diseases, which include Creutzfeldt-Jakob disease (CJD) and bovine spongiform encephalopathy (BSE), lead to severe neurological symptoms such as dementia, ataxia, and myoclonus. The exact mechanisms by which prions cause neurodegeneration are not fully understood, but recent studies suggest that prion infectivity and neurotoxicity may involve distinct pathways. Understanding these mechanisms is crucial for developing effective treatments for prion diseases and other neurodegenerative conditions.
4. Long incubation periods
Prion diseases are characterized by long incubation periods, which can span several years or even decades before clinical symptoms appear. During this silent phase, prions can accumulate in peripheral tissues and the central nervous system without causing overt disease. This prolonged incubation period poses significant challenges for early diagnosis and public health, as individuals may unknowingly transmit the disease during this time. The extended latency also complicates efforts to develop effective treatments, as interventions must target the disease before the onset of clinical symptoms.
5. Resistance to standard sterilization
Prions exhibit remarkable resistance to standard sterilization and decontamination procedures, making them particularly challenging to manage in medical and laboratory settings. Unlike conventional pathogens, prions are not easily inactivated by heat, radiation, or chemical disinfectants. This resistance has led to cases of iatrogenic transmission, where prion diseases are spread through contaminated medical instruments. Effective prion inactivation methods must be validated against specific prion strains to ensure their efficacy, as different strains may exhibit varying levels of resistance.
6. Multiple modes of transmission
Prion diseases can be transmitted through multiple routes, including sporadic, hereditary, and infectious forms. Sporadic cases arise without any known cause, while hereditary forms are linked to mutations in the prion protein gene (PRNP). Infectious transmission can occur through exposure to contaminated tissues, medical instruments, or consumption of prion-infected food products. The ability of prions to cross species barriers, as seen in the transmission of BSE to humans (variant CJD), underscores the importance of understanding the mechanisms of prion transmission and implementing effective control measures.
7. No effective treatments
Despite extensive research efforts, there are currently no effective treatments for prion diseases, and they remain uniformly fatal. Various therapeutic approaches, including small molecules, antibodies, and gene therapies, have been explored, but none have shown clear efficacy in clinical trials. The development of effective treatments is hindered by the complex and poorly understood mechanisms of prion propagation and neurotoxicity. Advances in early diagnosis and a better understanding of prion biology are essential for developing successful therapeutic strategies.
How Prions Work
Mechanism of Prion Infection
Prion infection is initiated when the normal cellular prion protein (PrPC) undergoes a conformational change to the pathogenic form, PrPSc. This process can be triggered by various factors, including prion infection, genetic mutations, and even viral infections such as influenza A virus. The conversion involves a decrease in α-helical content and an increase in β-strand content, which is facilitated by interactions with nucleic acids and glycosaminoglycans. These interactions act as scaffolds, promoting the misfolding and aggregation of PrPC into PrPSc, which then propagates the infectious state.
Conversion Process
The conversion of PrPC to PrPSc is a critical step in prion diseases and involves a complex interplay of molecular factors. The protein misfolding cyclic amplification (PMCA) technique has been instrumental in studying this process, revealing that specific RNA molecules and thiol-containing factors are essential for the conversion. Additionally, low pH conditions can trigger the conformational change, suggesting that the endocytic pathway and caveolae-like domains in cells may be involved in vivo. The conversion is also influenced by species-specific and polymorphic differences in the PrP sequence, which affect the efficiency and specificity of PrPSc formation.
Cascading Effect on Healthy Proteins
Once PrPSc is formed, it acts as a template to induce the misfolding of additional PrPC molecules, creating a cascading effect that amplifies the infectious agent. This process is highly efficient and can be modulated by various cellular factors, including plasmin, which has been shown to inhibit PrPSc formation by promoting α-cleavage of PrPC. The presence of specific RNA molecules and glycosaminoglycans further accelerates the conversion, leading to rapid propagation of the infectious state. This cascading effect is central to the pathogenesis of prion diseases, resulting in widespread neurodegeneration and associated clinical symptoms.
Types of human Prion Diseases
Human prion diseases are a group of rare, fatal brain disorders caused by misfolded prion proteins. These diseases lead to rapid neurodegeneration and are characterized by a variety of symptoms, depending on the specific type. Below are some of the main types of human prion diseases:

1. Creutzfeldt-Jakob Disease (CJD)
Creutzfeldt-Jakob Disease (CJD) is the most common form of human prion diseases, characterized by rapid neurodegeneration leading to dementia, myoclonus, and ultimately death. CJD can manifest in sporadic, familial, and iatrogenic forms. Sporadic CJD (sCJD) is the most prevalent, with no known cause, while familial CJD (fCJD) is linked to mutations in the prion protein gene (PRNP) on chromosome 20. Iatrogenic CJD results from medical procedures involving contaminated instruments or tissues. The disease is marked by the accumulation of an abnormal, protease-resistant prion protein (PrP^Sc) in the brain, leading to spongiform changes and neuronal loss.
2. Variant Creutzfeldt-Jakob Disease (vCJD)
Variant Creutzfeldt-Jakob Disease (vCJD) emerged in the UK in 1996 and is linked to the consumption of beef contaminated with bovine spongiform encephalopathy (BSE) prions. Unlike sporadic CJD, vCJD affects younger individuals and presents with psychiatric symptoms, followed by neurological decline. The disease is characterized by the presence of florid plaques in the brain, which are amyloid deposits surrounded by spongiform changes. The distinct histopathological features and the zoonotic origin of vCJD have significant public health implications, necessitating stringent measures to prevent transmission through the food supply.
3. Fatal Insomnia
Fatal Insomnia is a rare prion disease that can occur in both familial (FFI) and sporadic forms. FFI is caused by a specific mutation at codon 178 of the PRNP gene, coupled with a methionine at codon 129. The disease is characterized by severe insomnia, autonomic dysfunction, and rapid cognitive decline, with predominant pathological changes in the thalamus. Sporadic Fatal Insomnia (sFI) shares similar clinical and pathological features but occurs without a family history or identifiable genetic mutation. Both forms are invariably fatal, with death typically occurring within a year of symptom onset.
4. Gerstmann-Sträussler-Scheinker Syndrome (GSS)
Gerstmann-Sträussler-Scheinker Syndrome (GSS) is a hereditary prion disease associated with various mutations in the PRNP gene. It presents with a diverse clinical spectrum, including progressive cerebellar ataxia, spastic paraparesis, and dementia. Neuropathologically, GSS is marked by the presence of multicentric amyloid plaques and neurofibrillary tangles, which can overlap with features of Alzheimer’s disease. The disease progresses slowly over several years, and its clinical and pathological heterogeneity can complicate diagnosis. GSS is transmitted in an autosomal dominant pattern and is one of the first prion diseases linked to a specific genetic mutation.
5. Kuru
Kuru is a historically significant prion disease that was endemic among the Fore people of Papua New Guinea. It was transmitted through ritualistic endocannibalism, where individuals consumed the tissues of deceased relatives. Kuru primarily affected women and children and presented with progressive cerebellar ataxia, tremors, and eventual death. The cessation of these practices in the late 1950s led to a dramatic decline in Kuru cases. Neuropathologically, Kuru is characterized by spongiform changes, neuronal loss, and the presence of amyloid plaques in the brain. The study of Kuru has provided valuable insights into the transmission and pathogenesis of prion diseases.
Types of Animal Prion Diseases
Animal prion diseases are a group of fatal neurodegenerative disorders caused by misfolded prion proteins. These diseases affect various animal species and are characterized by progressive brain damage and behavioral changes. Below are some of the main types of animal prion diseases:

1. Scrapie
Scrapie is a prion disease that primarily affects sheep and goats, characterized by neurodegenerative symptoms such as ataxia, tremors, and intense itching, leading to scraping behavior. The disease is caused by the accumulation of abnormal prion proteins (PrPSc) in the brain, which leads to spongiform degeneration and neuronal loss. Scrapie has been endemic in sheep populations for many decades and is known to harbor mixtures of strains in different proportions, complicating its characterization and diagnosis. Recent studies have utilized transgenic mice overexpressing ovine prion protein to better understand the strain diversity and pathogenesis of scrapie.
2. Bovine Spongiform Encephalopathy (BSE) or “Mad Cow Disease”
Bovine Spongiform Encephalopathy (BSE), commonly known as “Mad Cow Disease,” is a prion disease that affects cattle. It is characterized by spongiform degeneration and astrocytosis in the brain, leading to severe neurological symptoms and death. BSE gained significant attention due to its zoonotic potential, as it has been linked to variant Creutzfeldt-Jakob disease (vCJD) in humans through the consumption of contaminated beef. The disease is caused by abnormal prion proteins and can be transmitted through contaminated meat and bone meal. Studies have shown that BSE prions can spread from the gut to the central nervous system via sensory nerve fibers.
3. Chronic Wasting Disease (CWD)
Chronic Wasting Disease (CWD) is a prion disease that affects deer, elk, and moose. It is characterized by weight loss, behavioral changes, and ultimately death. CWD is highly contagious among cervids and is transmitted through direct contact with infected animals or contaminated environments. The disease has been spreading rapidly in North America, raising concerns about its potential impact on wildlife populations and the risk of transmission to humans. The prion proteins responsible for CWD are similar to those causing other transmissible spongiform encephalopathies, but the mechanisms of interspecies transmission and the potential for human infection remain areas of active research.
4. Transmissible Mink Encephalopathy (TME)
Transmissible Mink Encephalopathy (TME) is a prion disease that affects farmed mink. It is characterized by behavioral changes, ataxia, and spongiform degeneration of the brain. TME is believed to be transmitted through the consumption of contaminated feed, similar to BSE in cattle. The disease has been experimentally transmitted to other species, including cattle and sheep, indicating the potential for cross-species transmission of prion diseases. Research on TME has provided valuable insights into the mechanisms of prion propagation and the factors influencing susceptibility to prion diseases.
5. Feline Spongiform Encephalopathy (FSE)
Feline Spongiform Encephalopathy (FSE) is a prion disease that affects domestic cats and captive wild felids. It is characterized by behavioral changes, ataxia, and spongiform degeneration of the brain. FSE was first identified in the 1990s and is believed to be linked to the consumption of BSE-contaminated meat products. The disease shares many pathological features with other prion diseases, including the accumulation of abnormal prion proteins in the brain. The occurrence of FSE highlights the potential for prion diseases to affect a wide range of species and underscores the importance of controlling prion contamination in the food supply.
Transmission of Prion Diseases
Prion diseases can be transmitted through various modes including genetic inheritance, consumption of contaminated food, and medical procedures. Genetic transmission occurs due to mutations in the prion protein gene, while consumption of contaminated food, such as beef infected with bovine spongiform encephalopathy (BSE), can lead to variant CJD. Medical procedures, particularly those involving neurosurgical instruments, have also been documented as a source of iatrogenic transmission due to the high resistance of prions to conventional sterilization methods. Prions exhibit an extraordinary resistance to standard sterilization techniques, including autoclaving and chemical disinfectants, which poses significant challenges for infection control in healthcare settings. This resistance is attributed to the prions’ ability to bind to surfaces of metal and plastic without losing infectivity, and their unusual thermostability, which allows them to survive even rigorous decontamination processes. Consequently, specialized prion-specific sterilization protocols, such as the use of sodium hydroxide or enzymatic treatments, are required to effectively inactivate prions on medical instruments.
Common symptoms of prion diseases
Prion diseases are characterized by a range of neurological and psychological symptoms that progressively worsen over time. These symptoms result from the accumulation of misfolded prion proteins in the brain, leading to severe neurodegeneration. Below are some common symptoms associated with prion diseases:

1. Memory Loss
Memory loss is a prominent symptom of prion diseases, often manifesting early in the disease progression. This symptom is primarily due to the neurodegenerative nature of prion diseases, which cause significant damage to brain tissues, leading to cognitive decline and dementia. Patients with prion diseases, such as Creutzfeldt-Jakob disease (CJD), frequently experience rapid and severe memory impairment, which can be one of the first signs that prompt medical evaluation. The progressive nature of memory loss in prion diseases typically worsens over time, contributing to the overall decline in intellectual function and quality of life.
2. Confusion
Confusion is another common symptom associated with prion diseases, often accompanying memory loss and other cognitive impairments. This confusion can manifest as disorientation, difficulty in understanding or processing information, and an inability to perform routine tasks. The rapid progression of prion diseases exacerbates these symptoms, leading to a significant decline in mental clarity and cognitive function. Patients may struggle with recognizing familiar people or places, further complicating their daily lives and increasing their dependence on caregivers.
3. Difficulty Walking and Changes in Gait
Difficulty walking and changes in gait are hallmark symptoms of prion diseases, often resulting from the neurodegenerative impact on the cerebellum and other parts of the brain responsible for motor control. Patients may exhibit ataxia, characterized by unsteady and uncoordinated movements, which can lead to frequent falls and mobility issues. These motor symptoms typically worsen as the disease progresses, severely affecting the patient’s ability to perform daily activities independently. Early recognition of these symptoms can aid in the timely diagnosis of prion diseases.
4. Loss of Coordination (Ataxia)
Loss of coordination, or ataxia, is a significant symptom of prion diseases, reflecting the extensive damage to the brain’s motor control regions. Ataxia manifests as clumsiness, unsteady movements, and difficulty with tasks requiring fine motor skills. This symptom is often one of the earliest and most noticeable signs of prion diseases, such as CJD, and can rapidly progress to severe motor impairment. The presence of ataxia, along with other neurological symptoms, can help differentiate prion diseases from other neurodegenerative conditions.
5. Vision Problems
Vision problems are frequently observed in patients with prion diseases, often resulting from the degeneration of neural pathways involved in visual processing. These issues can include blurred vision, double vision, and difficulty with visual coordination. In some cases, patients may experience visual hallucinations, further complicating their condition. The progressive nature of prion diseases means that vision problems can worsen over time, contributing to the overall decline in the patient’s functional abilities and quality of life.
6. Hallucinations
Hallucinations, particularly visual hallucinations, are common psychiatric manifestations of prion diseases. These hallucinations can occur early in the disease course and are often accompanied by other neuropsychiatric symptoms such as delusions and agitation. The presence of hallucinations can be distressing for both patients and caregivers, complicating the management of the disease. Recognizing these symptoms can aid in the early diagnosis and differentiation of prion diseases from other neurodegenerative disorders.
7. Anxiety and Depression
Anxiety and depression are prevalent psychiatric symptoms in patients with prion diseases, often appearing in the early stages of the illness. These affective symptoms can significantly impact the patient’s quality of life and may precede more severe neurological manifestations. The rapid progression of prion diseases can exacerbate these psychiatric symptoms, leading to increased emotional distress and a greater need for psychological support and intervention. Early identification and management of anxiety and depression are crucial for improving patient outcomes.
8. Fatigue
Fatigue is a common symptom in prion diseases, reflecting the overall decline in physical and cognitive function. Patients often experience profound tiredness and a lack of energy, which can interfere with daily activities and reduce their ability to engage in social interactions. The neurodegenerative nature of prion diseases contributes to this fatigue, as the brain’s ability to regulate and sustain normal bodily functions deteriorates over time. Addressing fatigue through supportive care and symptom management is essential for maintaining the patient’s quality of life.
Diagnosis of Prion Diseases
Diagnosing prion diseases involves a combination of clinical evaluation, imaging studies, and laboratory tests. Due to the rarity and complexity of these diseases, a definitive diagnosis often requires specialized techniques. Below are some common methods used in the diagnosis of prion diseases:
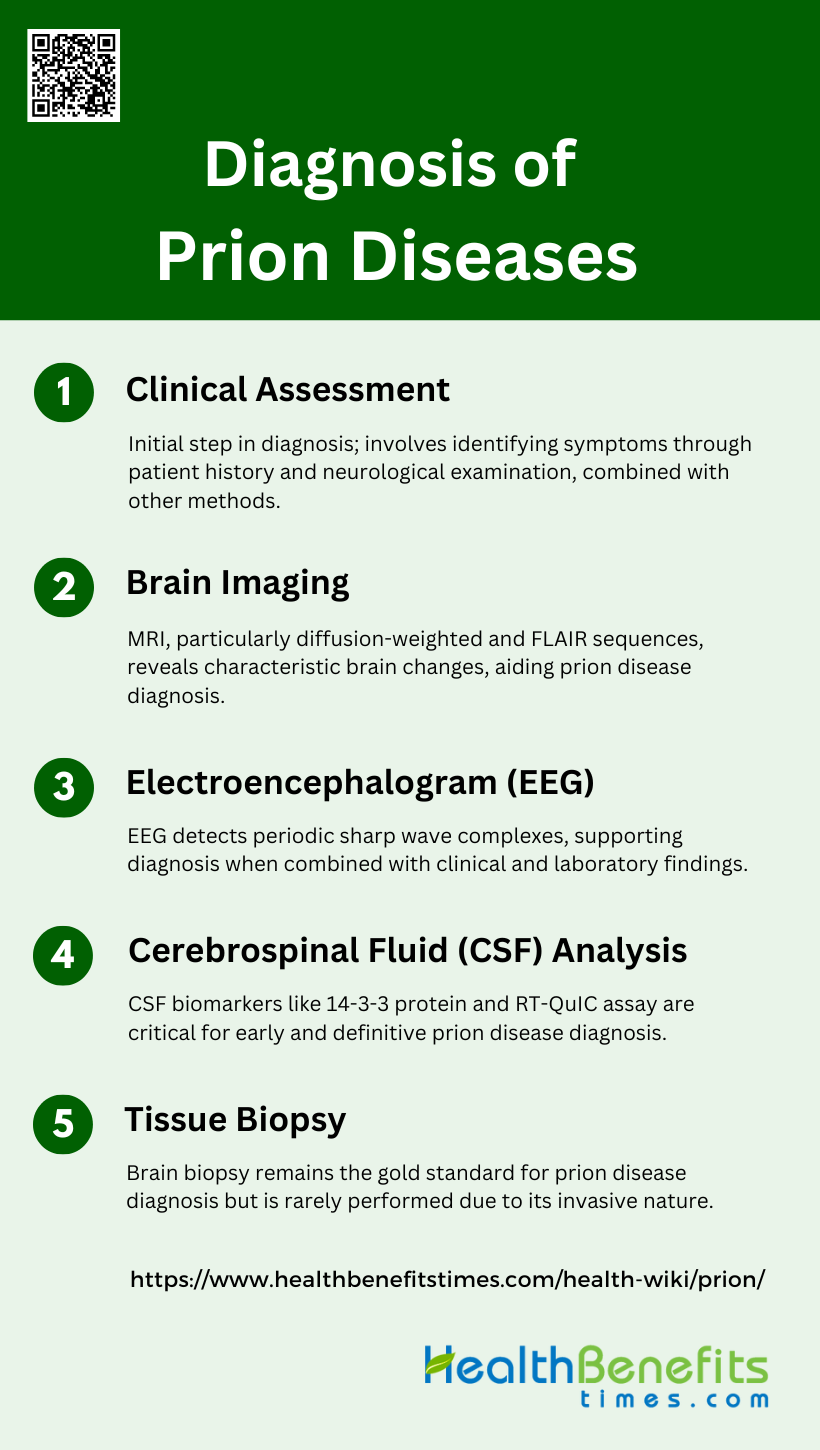
1. Clinical Assessment
Clinical assessment is the initial step in diagnosing prion diseases, which are rapidly progressive and fatal neurodegenerative disorders. The clinical spectrum of prion diseases is varied, but common symptoms include rapidly progressive dementia, cerebellar ataxia, and myoclonus. Clinical evaluation often involves a detailed patient history and neurological examination to identify these symptoms. However, clinical assessment alone is not sufficient for a definitive diagnosis due to the overlap of symptoms with other neurodegenerative diseases. Therefore, clinical assessment is typically combined with other diagnostic methods to improve accuracy.
2. Brain Imaging
Brain imaging, particularly magnetic resonance imaging (MRI), plays a crucial role in the diagnosis of prion diseases. Diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences are particularly useful. These imaging techniques can reveal characteristic changes in the brain, such as hyperintensities in the basal ganglia and cortical regions, which are indicative of prion diseases like Creutzfeldt-Jakob disease (CJD). Recent studies have shown that MRI with diffusion-weighted images has high sensitivity and specificity for diagnosing sporadic CJD, making it a reliable non-invasive diagnostic tool.
3. Electroencephalogram (EEG)
Electroencephalogram (EEG) is another important diagnostic tool for prion diseases. EEG can detect periodic sharp wave complexes, which are characteristic of prion diseases such as CJD. Although not specific to prion diseases, the presence of these periodic discharges can support the diagnosis when combined with clinical and other laboratory findings. EEG is particularly useful in differentiating prion diseases from other causes of rapidly progressive dementia.
4. Cerebrospinal Fluid (CSF) Analysis
Cerebrospinal fluid (CSF) analysis is a critical component in the diagnosis of prion diseases. Biomarkers such as 14-3-3 protein, tau protein, and neuron-specific enolase (NSE) are commonly elevated in the CSF of patients with prion diseases. The real-time quaking-induced conversion (RT-QuIC) assay has emerged as a highly sensitive and specific test for detecting prion protein in the CSF. RT-QuIC has been incorporated into diagnostic criteria due to its high diagnostic accuracy, making it a valuable tool for early and definitive diagnosis.
5. Tissue Biopsy
Tissue biopsy, particularly brain biopsy, remains the gold standard for the definitive diagnosis of prion diseases. Histopathological examination of brain tissue can reveal the presence of abnormal prion protein deposits and spongiform changes characteristic of prion diseases. However, due to its invasive nature, brain biopsy is rarely performed antemortem and is usually reserved for cases where other diagnostic methods are inconclusive. Advances in less invasive techniques, such as RT-QuIC, have reduced the reliance on brain biopsy for diagnosis.
Treatment and Management of Prion Diseases
Managing prion diseases involves a multifaceted approach aimed at alleviating symptoms and improving the patient’s quality of life. While there is no cure, various strategies can help manage the disease’s progression. Below are some key aspects of treatment and management for prion diseases:

1. Supportive Care
Supportive care for prion diseases focuses on maintaining the patient’s comfort and quality of life, as there is no cure for these rapidly progressive neurodegenerative conditions. This type of care often involves a multidisciplinary approach, including neurologists, palliative care specialists, and social workers, to address the complex needs of the patient and their family. Supportive care strategies are similar to those used in geriatric and hospice care, emphasizing symptom management, psychosocial support, and education for caregivers. The National Prion Clinic, for example, provides comprehensive care and facilitates research to improve early diagnosis and potential therapies.
2. Symptom Management
Symptom management in prion diseases is crucial due to the rapid progression and severe neurological decline associated with these conditions. Pharmacologic treatments may include medications to manage pain, seizures, and psychiatric symptoms such as anxiety and depression. Nonpharmacologic strategies, such as physical therapy and cognitive exercises, can also be beneficial. The goal is to alleviate symptoms and improve the patient’s quality of life as much as possible. Additionally, monitoring tools have been developed to track symptom progression and adjust care plans accordingly, which is essential for both patients and caregivers.
3. Assistance with Daily Activities
Assistance with daily activities is a significant aspect of managing prion diseases, as patients often experience severe cognitive and motor impairments. Caregivers play a vital role in helping with tasks such as bathing, dressing, eating, and mobility. The burden on caregivers can be substantial, leading to burnout and the need for additional support services. Structured care plans and respite care options are essential to ensure that both patients and caregivers receive the necessary support. Education and training for caregivers are also critical to help them manage the unique challenges posed by prion diseases.
4. Nutrition and Hydration
Maintaining proper nutrition and hydration is challenging in prion diseases due to difficulties with swallowing and reduced appetite. Nutritional support may involve the use of high-calorie supplements, modified food textures, and, in some cases, feeding tubes to ensure adequate intake. Hydration is equally important and may require regular monitoring and assistance with fluid intake. Addressing these needs is vital to prevent complications such as malnutrition and dehydration, which can exacerbate the patient’s condition and reduce their quality of life. Care teams often include dietitians to develop individualized nutrition plans tailored to the patient’s needs.
5. Experimental Approaches
Experimental approaches to treating prion diseases are an active area of research, given the lack of effective therapies. Various strategies are being explored, including chemical compounds, immunotherapy, and gene-silencing techniques targeting the prion protein. Recent studies have shown some promise in animal models, such as the use of heterologous prion proteins to inhibit disease progression. Systems biology approaches have also identified potential therapeutic targets by mapping gene expression changes associated with prion diseases. While these experimental treatments are still in the early stages, they offer hope for future therapeutic options.
Prevention of Prion Diseases
Preventing prion diseases requires a comprehensive approach that includes dietary precautions, stringent regulations, and public health measures. These strategies aim to minimize the risk of transmission and ensure the safety of both humans and animals. Below are some key methods for preventing prion diseases:

1. Avoid Consuming Infected Animals
Avoiding the consumption of infected animals is a critical measure in preventing prion diseases. The transmission of prion diseases such as variant Creutzfeldt-Jakob disease (vCJD) has been linked to the ingestion of contaminated meat, particularly during the BSE (mad cow disease) epidemic in the 1980s. Surveillance programs have revealed that prion diseases in ruminant populations were previously underestimated, highlighting the importance of strict food safety measures. By avoiding the consumption of potentially infected animals, the risk of zoonotic transmission of prion diseases to humans can be significantly reduced.
2. Food Safety Regulations
Implementing stringent food safety regulations is essential to prevent the spread of prion diseases. Following the BSE crisis, large-scale epidemiological surveillance programs were established in the EU to monitor and control prion diseases in livestock. These regulations include banning the use of animal by-products in feed, rigorous testing of cattle, and the removal of specified risk materials from the food chain. Such measures have been effective in reducing the incidence of prion diseases and protecting public health by minimizing the risk of contaminated food entering the market.
3. Decontamination of Medical Instruments
Decontaminating medical instruments is crucial to prevent iatrogenic transmission of prion diseases. Prions are highly resistant to conventional sterilization methods, and standard decontamination procedures may not be sufficient to eliminate prion infectivity. The CDC and WHO have developed specific guidelines for the decontamination of prion-contaminated instruments, emphasizing the need for prion-specific disinfection protocols. Ensuring that medical instruments are properly decontaminated can significantly reduce the risk of prion transmission during surgical and medical procedures.
4. Genetic Testing and Counseling
Genetic testing and counseling play a vital role in managing the risk of prion diseases, particularly for individuals with a family history of these conditions. Some prion diseases, such as familial Creutzfeldt-Jakob disease, are linked to genetic mutations in the prion protein gene. Identifying individuals who carry these mutations through genetic testing can help in providing appropriate counseling and preventive measures. Early detection and intervention can potentially delay the onset of symptoms and improve the quality of life for those at risk.
5. Surveillance and Testing
Surveillance and testing are fundamental components in the prevention and control of prion diseases. Continuous monitoring of both animal and human populations is necessary to detect and respond to prion disease outbreaks promptly. Surveillance programs have been instrumental in identifying new forms of prion diseases and assessing their zoonotic potential. Regular testing of livestock, particularly in regions with a history of prion diseases, helps in early detection and containment, thereby preventing the spread of these fatal neurodegenerative diseases.
6. Preventing Iatrogenic Transmission
Preventing iatrogenic transmission of prion diseases involves implementing strict infection control measures in healthcare settings. Prions can be transmitted through contaminated medical instruments, blood products, and tissues. Adhering to prion-specific decontamination protocols and using disposable instruments when possible can minimize the risk of transmission. Additionally, healthcare workers should be educated about the risks and prevention strategies associated with prion diseases to ensure compliance with safety guidelines.
7. Public Health Measures
Public health measures are essential to mitigate the risk of prion diseases. These measures include public education campaigns to raise public awareness about prion diseases and their transmission routes, as well as the implementation of policies to control the spread of prions in both animal and human populations. Governments and health organizations must collaborate to develop and enforce regulations that protect public health, such as banning high-risk practices like the use of animal by-products in feed and ensuring the safe disposal of prion-contaminated materials.
Why Are Prions So Unique and Dangerous?
Prions are uniquely dangerous proteins that challenge our understanding of infectious agents. Unlike viruses or bacteria, prions contain no genetic material, consisting solely of misfolded proteins that can trigger a cascade of protein misfolding in the brain. This unique structure makes prions incredibly resistant to conventional sterilization methods, allowing them to persist in the environment and spread between organisms with alarming efficiency. Prions cause fatal neurodegenerative disorders like mad cow disease by inducing normal proteins to misfold and aggregate, leading to progressive brain damage. Their ability to propagate without nucleic acids and their extreme resistance to destruction make prions particularly challenging for medical science and public health, as traditional approaches to preventing and treating infectious diseases are often ineffective against these insidious agents.
Misconception about Prions
Prions are often misunderstood due to their unique nature and the complexity of the diseases they cause. These misconceptions can lead to unnecessary fear or inadequate precautions. Below are some common misconceptions about prions:

1. Prions are living organisms
Contrary to popular belief, prions are not living organisms. They are misfolded proteins that lack any genetic material such as DNA or RNA, which are essential components of living entities. Prions are unique infectious agents composed solely of proteins, specifically abnormal forms of the prion protein (PrP). Their ability to propagate and cause disease stems from their capacity to induce normal proteins to misfold and aggregate, rather than from any metabolic processes associated with living organisms. This fundamental misunderstanding often leads to confusion about how prions function and why they are so challenging to combat.
2. Prions can be easily killed or deactivated
The notion that prions can be easily killed or deactivated is a dangerous misconception. Unlike most pathogens, prions are extraordinarily resistant to conventional sterilization methods. Their unique protein-only structure makes them impervious to many standard decontamination procedures that typically destroy other infectious agents. Prions can withstand high temperatures, radiation, and chemical treatments that would normally inactivate viruses or bacteria. This extreme resilience poses significant challenges for medical professionals and researchers in preventing the spread of prion diseases and decontaminating infected materials.
3. Prion diseases are highly contagious
While prion diseases are transmissible, they are not highly contagious in the way that many people assume. Prion diseases are extremely rare, occurring in about 1 in a million people. Unlike common infectious diseases, prion diseases are not spread through casual contact, airborne transmission, or typical person-to-person interactions. The primary modes of transmission are through consumption of infected tissue, exposure to contaminated medical instruments, or genetic inheritance. However, certain prion diseases like kuru and variant Creutzfeldt-Jakob disease (vCJD) have shown the potential for transmission through specific practices or contaminated food sources.
4. Prion diseases only affect the elderly
It is a misconception that prion diseases exclusively affect the elderly. While it’s true that some forms of prion diseases, such as sporadic Creutzfeldt-Jakob disease (sCJD), are more common in older adults, prion diseases can affect individuals of all ages. Genetic forms of prion diseases, like familial CJD or Gerstmann-Sträussler-Scheinker syndrome, can manifest at younger ages. Additionally, acquired forms of prion diseases, such as variant CJD linked to bovine spongiform encephalopathy (BSE), have affected younger individuals. The age of onset can vary widely depending on the specific type of prion disease and the mode of transmission.
5. Prions only cause brain diseases
While prion diseases are primarily known for their devastating effects on the brain, it is incorrect to assume that prions only cause brain diseases. Recent research has shown that prions can affect other parts of the body as well. Although the most prominent and fatal effects are indeed in the central nervous system, prions can accumulate in various tissues and organs throughout the body. This broader distribution of prions has implications for disease transmission and diagnosis. Furthermore, the concept of prion-like behavior has been extended to other protein misfolding disorders affecting different body systems, challenging the notion that prions are exclusively associated with brain diseases.
FAQs
1. What are the differences between prion diseases and other neurodegenerative diseases like Alzheimer’s or Parkinson’s?
Prion diseases are unique in that they are caused by infectious misfolded proteins (prions) that can propagate themselves by converting normal proteins into the same misfolded form. While Alzheimer’s and Parkinson’s diseases also involve protein misfolding and aggregation, they are not typically infectious or transmissible between individuals.
2. How do prion diseases spread in animal populations, particularly in the wild?
Prion diseases like Chronic Wasting Disease (CWD) in deer and elk spread through direct animal-to-animal contact, as well as indirectly through contaminated environments (e.g., soil, water, or vegetation). Prions can remain infectious in the environment for years, contributing to the disease’s persistence and spread among wild populations.
3. Can prion diseases be detected before symptoms appear?
Currently, there are no reliable methods to detect prion diseases before clinical symptoms appear. However, research is ongoing to develop biomarkers and diagnostic tools, such as advanced imaging techniques and blood tests, that could potentially identify prion diseases in their early stages.
4. Why do prion diseases have such long incubation periods?
The long incubation periods of prion diseases are not fully understood but are believed to be due to the slow accumulation of misfolded prion proteins in the brain and other tissues. During this silent phase, prions may not reach sufficient levels to cause noticeable symptoms, allowing the disease to go undetected for years or even decades.
5. Is there a risk of prion disease transmission through blood transfusions or organ transplants?
Yes, there is a potential risk of transmitting prion diseases through blood transfusions or organ transplants from donors who have undiagnosed prion diseases. However, this risk is considered low, and blood banks and transplant centers have implemented strict screening protocols to minimize it.
6. Can prions be present in the environment, and if so, how long can they remain infectious?
Yes, prions can be present in the environment, particularly in soil and water contaminated by infected animal tissues or bodily fluids. Prions are highly stable and can remain infectious in the environment for several years, making it challenging to control prion disease outbreaks in wildlife.
7. What precautions should hunters take to avoid exposure to prion diseases like Chronic Wasting Disease (CWD)?
Hunters should follow guidelines for handling and processing deer, elk, and moose, especially in areas where CWD is known to occur. This includes wearing gloves, avoiding contact with brain or spinal tissue, and having meat tested for CWD before consumption. It’s also advisable to avoid consuming meat from animals that appear sick.
8. Are there any known ways to decontaminate environments that have been exposed to prions?
Decontaminating environments exposed to prions is extremely challenging due to their resistance to conventional disinfectants and environmental stability. Current methods include the use of strong chemical treatments, such as sodium hypochlorite or sodium hydroxide, and incineration at high temperatures. However, these methods are not always practical for large areas.
9. Can animals other than humans and livestock be affected by prion diseases?
Yes, prion diseases can affect a variety of animals beyond humans and livestock. For example, Chronic Wasting Disease affects deer, elk, and moose, while Transmissible Mink Encephalopathy (TME) affects farmed mink, and Feline Spongiform Encephalopathy (FSE) affects domestic and wild felids.
10. Are prion diseases considered a bioterrorism threat?
Prions are considered a potential bioterrorism threat due to their highly infectious nature, resistance to decontamination, and the devastating effects of prion diseases. However, the practical use of prions for bioterrorism is limited by challenges in dissemination and targeting.